病史摘要
男,63岁,5天前突发精神行为异常,撕坏、打碎家里物品,否认基础慢性病,无特殊药物服用史,无明显诱因,当天有低热,37.8度左右;否认手术外伤史、否认高血压、糖尿病史。
MR影像图




MRS

MR平扫+DWI+MRS:双侧额叶、半卵圆中心、侧脑室旁、胼胝体白质区见片状稍长T1稍长T2信号,FLAIR像显示高信号,边缘模糊,DWI像上病灶边缘见条片状高信号,ADC图上呈稍低信号,MRS示:上述病灶CHO波未见增高,NAA波未见减低,CHO/NAA为0.58。
诊断:双侧额叶、半卵圆中心、侧脑室旁、胼胝体区对称性脑白质脱髓鞘,考虑神经元核内包涵体病可能。
随访




病例分析
分析:老年男性,本次发病呈急性发作。影像表现:皮髓质交界区广泛DWI鸡冠花样、曲线样高信号、胼胝体DWI高信号、对称性脑白质病变、皮层肿胀、广泛性脑萎缩。
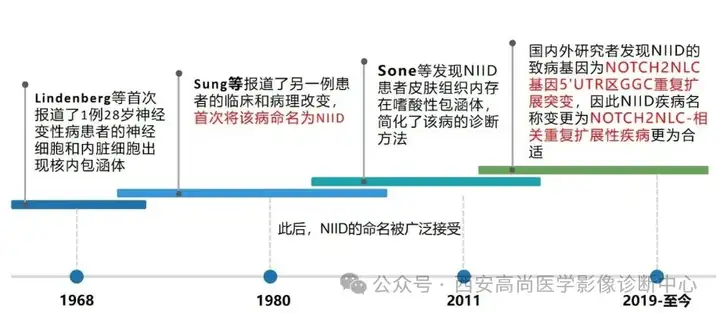
神经元核内包涵体病(NIID)
01 概述
神经元核内包涵体病(NIID)是一种主要累及神经系统的罕见遗传性疾病,其病理特征为在多种系统的组织细胞中出现嗜酸性的核内包涵体。
NIID的致病突变为NOTCH2NLC基因的GGC重复扩展同时发现该基因变异除了导致以发作性/进展性脑病、周围神经病和自主神经病等为主要表现的经典型NIID外,在少数AD、PD、MS、ET等神经系统变性病患者中也检测到此基因变异。因此NIID疾病名称变更为NOTCH2NLC-相关重复扩展性疾病更为合适。


成人型NIID临床表现
发病年龄:存在2个高峰期。30岁左右发病者以肢体无力为主要表现;55岁左右发病者以认知障碍为主要表现。
中枢神经系统症状:发作性脑病、排尿障碍、肢体震颤、小脑性共济失调、帕金森综合征、偏头痛发作、视力减退。
发作性脑病具有很强的诊断提示价值,表现为突发的意识障碍、意识淡漠、谵妄、精神症状、虚弱等,发生率从18%-64.5%不等。
自主神经功能障碍:部分患者可出现不明原因尿潴留或尿失禁,可以早于其他症状多年出现。其他自主神经功能障碍:双侧瞳孔缩小、胃肠道功能紊乱、体位性低血压、心律失常、性功能障碍等。
患者可以表现为反复普通型偏头痛为首发症状、快速进展的痴呆、反复发作的共济失调、反复发作的Miller-Fisher综合征、肢体无力合并眼外肌麻痹。这些临床表型具有相当的不典型性,易被漏诊或误诊。
02 病理特点
广泛存在于中枢、周围、自主神经系统神经元细胞核内的嗜酸性透明包涵体;光镜下可见此包涵体邻近核仁,呈圆形、直径1.5-10um,HE染色呈嗜酸性。免疫组化染色可见泛素和p62阳性,电镜下可见其由无膜结构的纤维物质组成。包涵体不仅可出现在神经元内,也可出现在胶质细胞、心肌细胞、骨骼肌细胞、肾小管上皮细胞、施万细胞、汗腺导管上皮细胞、成纤维细胞、脂肪细胞中。
NIID是一个病理学诊断,因此尽管目前有NIID的基因检查,仍需病理上发现核内包涵体方可诊断。皮肤活检位置:外踝上方10cm处皮肤组织。
嗜酸性核内包涵体也可在其他 GGC 重复扩张疾病也可观察到,包括脆性X相关震颤/共济失调综合症(FXTAS)和眼远端肌病(OPDM)。据报道FXTAS患者大脑和外周的神经元和星形胶质细胞中广泛检测到泛素阳性核内包涵体,其大小、形状、成分和分布与NIID相似。
新开发的针对 uN2CpolyG 的抗体,可能有助于NID的识别,需要在NIID 患者的组织中进一步验证。
2019年研究者发现NIID的致病基因为NOTCH2NLC基因5'UTR区GGC重复扩展突变后,研究均表明,该基因突变不仅可以导致以发作性或进展性脑病、周围神经病和自主神经病等为核心症状的经典型ND,而且在少数AD、额颞叶痴呆、PD、MS、ET、成年型白质脑病等神经系统变性病患者中也可被检测到。
NIID属于一组三核苷酸重复疾病(TRD),具有一些独特的遗传特征,包括重复大小的致病阈值的存在、重复大小与发病年龄之间的相关性、重复大小与临床表型之间的相关性、性别偏倚等。

03 影像表现
(1)皮髓质交界区曲线样DWI高信号
为从额顶颞叶皮髓质交界区开始,随着病情发展向大脑后部延伸,形成皮质下“鸡冠花样”或“绸带征”的DWI高信号(具有一定的特异性,病理证实为海绵样变)。家系NID患者仅有37.5%出现上述DWI改变。
(2)胼胝体DWI高信号
部分患者在病程早期仅出现胼胝体的DWI高信号,提示胼胝体联络纤维和皮质下弓形纤维具有类似的易受累性。需要和可逆性胼胝体压部脑病、Marchiafava-Bignami综合征进行鉴别。
(3)对称性脑白质病变
显著累及放射冠和半卵圆中心部位的白质病灶。
(4)皮质肿胀和增强
部分患者在病程的某些阶段出现沿皮质分布的DWI和FLAIR高信号,常伴随沿皮质表面的线样强化(可能与亚急性炎症发作相关),通常分布在后部皮质,且多在临床急性发作期出现,因而要注意和MELAS鉴别。
(5)广泛的脑萎缩和沿小脑中轴对称分布的小脑白质病变
影像表现新进展
DWI异常信号可在主要症状出现数年后出现,甚至可能随着疾病进展而完全消失。
皮质髓质交界处DWI高信号曾被认为是NID的影像标志,但在IID的肌无力表型和帕金森病表型中却非常少见。
MRI阴性结果不能作为NIID的排除标准,建议长期随访或复查。
NIID的临床诊断流程与思路

04 鉴别诊断

伴皮质下和白质脑病的常染色体显性遗传性脑动脉病(CADASIL):是一种成年发病的显性遗传性小血管病,致病基因定位于19p12的Notch3基因.影像上常表现头白质高信号、腔隙性梗死、脑微出血。特征性的颞极白质高信号(0’Sulliva征)和外囊高信号(“人”字征)是诊断CADASIL影像学标志。

MELAS:本病为母系线粒体遗传性疾病,发病年龄10-40岁,临床上以卒中样发作MELAS:为特点,病灶主要累及大脑后半部的颞枕叶皮层或皮层下白质,病灶具有对称性游走性,急性期区域多呈高灌注,MRS倒置乳酸峰具有高度敏感性,多伴有脑萎缩改变。

克雅氏病:又称皮质-纹状体-脊髓变性。临床上以快速进行性痴呆、肌阵挛、共济失调为主要表现。典型影像学表现为对称或非对称性皮层和(或)基底节区受累,通常不累及中央回,扣带回、角回、楔前叶、额上回及纹状体为主要受累部位,DWI信号随病情进展信号增高,可持续数周,晚期消失,晚期脑萎缩改变。

皮层下动脉硬化性脑病(Binswanger病):本病为老年人在脑动脉硬化基础上,大脑半球白质弥漫性脱髓鞘性脑病。影像上表现为脑室周围白质与半卵圆中心显示散在或融合性病变区,T1WI呈低信号,T2WI呈高信号。重度与中度显示脑室扩大,腔隙性脑梗死显影清晰,FLAIR上脱髓鞘斑块及腔隙性脑梗死表现为高信号常伴有脑萎缩。

参考文献
1. XiuRong H ,BeiSha T ,Peng J, et al. The Phenotypes and Mechanisms of NOTCH2NLC-Related GGC Repeat Expansion Disorders: a Comprehensive Review.[J]. Molecularneurobiology,2021,59(1).
2. 洪道俊,王朝霞.神经元核内包涵体病的再认识[J].中华神经科杂志,2020,53(10):741-5745.DOI:10.3760/cma.j.cn113694-20200422-00291.
3. Cardoso D F,Mota DL A F G,Torres F P, et al. Imaging of Creutzfeldt-Jakob Disease:Imaging Patterns and Their Differential Diagnosis.[J]. Radiographics : a reviewpublication of the Radiological Society of North America, Inc, 2017, 37(1).
4. Lei B, Dandan Z,Qingjie l, et al. Current advances in neuronal intranuclearinclusion disease,, Neurological sciences : official journal of the ItalianNeurological Society and of the Italian Society of ClinicalNeurophysiology,2023,44(6).
